
Analyse des séquences
Analyses effectuées sur les données du CQDG
| Étude | WGS ou WES | Nombre de séquences disponibles dans l'étude | Nombre de séquences disponibles sur le portail | Données brutes soumises au portail | Pipeline Pre-processing / Alignement | Pipeline Calling | Pipeline Post-processing | Pipeline annotation | Fichiers disponibles sur le portail | Prochaines étapes |
|---|---|---|---|---|---|---|---|---|---|---|
| DEE | WGS | 588 | 588 | .gvcf / .cram / .cnv.vcf / .sv.vcf | Dragen | Dragen | Ferlab | Ferlab | Données brutes | |
| Cartagene | WGS | 2184 | 2184 | .gvcf / .cram / .cnv.vcf / .sv.vcf | Dragen | Dragen | Ferlab | Ferlab | Données brutes | |
| BACQ | WES | 466 | 465 | FastQ / .cram / .vcf (snv) | nf-core/Sarek | nf-core/Sarek | Ferlab | Ferlab | .cram / .vcf | |
| NeuroDev | WGS | 382 | 364 | FastQ / .cram / .vcf (snv) | nf-core/Sarek | nf-core/Sarek | Ferlab | Ferlab | .cram / .vcf | |
| PRAGMatiQ | WGS | 257 | 110 | FastQ / .cram / .vcf (snv) | nf-core/Sarek | nf-core/Sarek | Ferlab | Ferlab | .cram / .vcf |
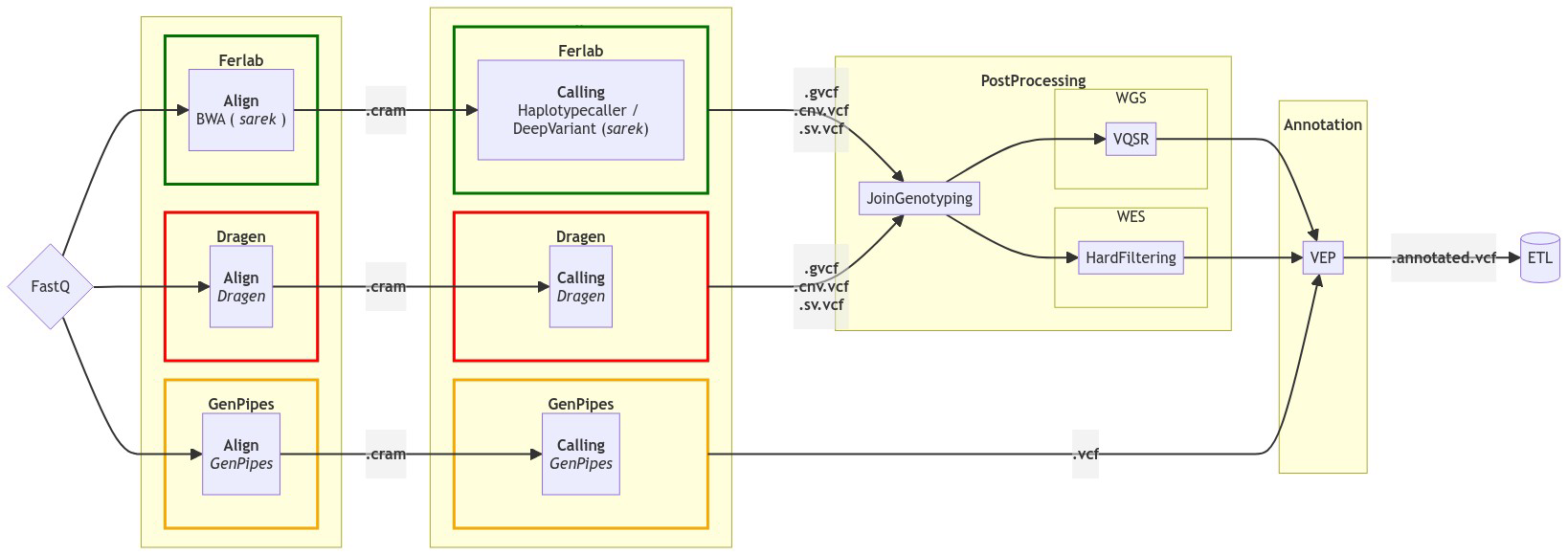
Description des pipelines bioinformatiques utilisés
Outils
GenPipes
GenPipes DNAseq Exome
Dragen
Filtre :
Hard filtering sur critère bas QUAL > 3, DP > 1
Ferlab
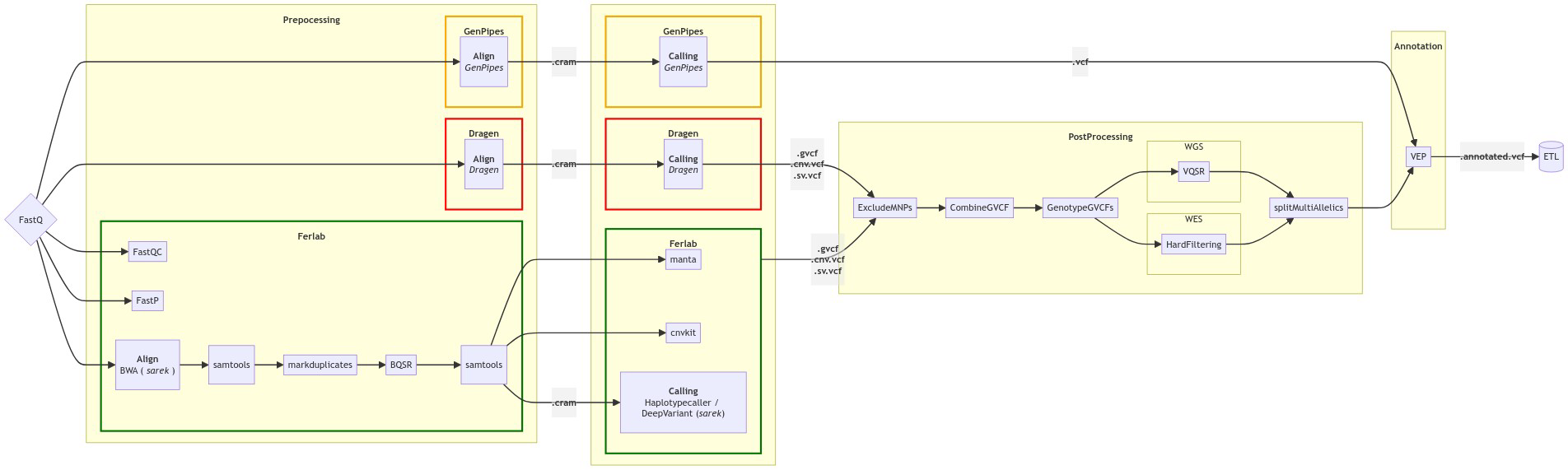
PreProcessing
Pour le PreProcessing nous utilisons le pipeline Sarek développé par la communauté nf-core.
Parmi les outils disponibles nous avons utilisé :
- des outils de contrôle de qualité
- FastQC
- FastP
- samtools
- un outil d'alignement
- BWA
- un outil afin de tagger les duplicatas de PCR
- markduplicates
- un outil pour la recalibration de la qualité des bases
- BQSR
Calling
Pour le calling nous avons choisi plusieurs outils parmi les outils proposés par Sarek
- Calling
- Haplotypecaller
- Deepvariant
PostProcessing
Dans l'étape de PostProcessing nous utilisons un pipeline que nous avons développé à l'interne à Ferlab.
- Exclusion des MNPs
- bcftools
- CombineGVCF par famille
- gatk
- GenotypeGVCFs calculate genotype
- gatk
- VQSR
- gatk vqsr
- splitMultiAllelics
- bcftools
Annotation
- VEP
- tabix
Version des logiciels
GenPipes
| Process Name | Software | Version |
|---|---|---|
| GenPipes DNAseq Exome | 4.1.2 | |
| gatk | 3.8 |
Dragen
Ferlab
Sarek + Ferlab
| Process Name | Software | Version |
|---|---|---|
| BCFTOOLS_MPILEUP | bcftools | '1.17' |
| BCFTOOLS_SORT | bcftools | '1.17' |
| BCFTOOLS_STATS | bcftools | '1.17' |
| BWAMEM1_MEM | bwa | 0.7.17-r1188 |
| BWAMEM1_MEM | samtools | 1.16.1 |
| CNNSCOREVARIANTS | gatk4 | 4.4.0.0 |
| CNVKIT_ANTITARGET | cnvkit | 0.9.10 |
| CNVKIT_BATCH | samtools | '1.17' |
| CNVKIT_GENEMETRICS | cnvkit | 0.9.10 |
| CNVKIT_REFERENCE | cnvkit | 0.9.10 |
| CREATE_INTERVALS_BED | gawk | 5.1.0 |
| CUSTOM_DUMPSOFTWAREVERSIONS | python | 3.12.0 |
| CUSTOM_DUMPSOFTWAREVERSIONS | yaml | 6.0.1 |
| DEEPVARIANT | deepvariant | 1.5.0 |
| FASTP | fastp | 0.23.4 |
| FASTQC | fastqc | 0.12.1 |
| FILTERVARIANTTRANCHES | gatk4 | 4.4.0.0 |
| FREEBAYES | freebayes | 1.3.6 |
| GATK4_APPLYBQSR | gatk4 | 4.4.0.0 |
| GATK4_BASERECALIBRATOR | gatk4 | 4.4.0.0 |
| GATK4_GATHERBQSRREPORTS | gatk4 | 4.4.0.0 |
| GATK4_HAPLOTYPECALLER | gatk4 | 4.4.0.0 |
| GATK4_MARKDUPLICATES | gatk4 | 4.4.0.0 |
| GATK4_MARKDUPLICATES | samtools | '1.17' |
| INDEX_CRAM | samtools | '1.17' |
| MANTA_GERMLINE | manta | 1.6.0 |
| MERGE_BCFTOOLS_MPILEUP | gatk4 | 4.4.0.0 |
| MERGE_CRAM | samtools | '1.17' |
| MERGE_DEEPVARIANT_GVCF | gatk4 | 4.4.0.0 |
| MERGE_DEEPVARIANT_VCF | gatk4 | 4.4.0.0 |
| MERGE_FREEBAYES | gatk4 | 4.4.0.0 |
| MERGE_HAPLOTYPECALLER | gatk4 | 4.4.0.0 |
| MERGE_STRELKA | gatk4 | 4.4.0.0 |
| MERGE_STRELKA_GENOME | gatk4 | 4.4.0.0 |
| MOSDEPTH | mosdepth | 0.3.3 |
| SAMTOOLS_STATS | samtools | '1.17' |
| SNPEFF_SNPEFF | snpeff | 5.1d |
| STRELKA_SINGLE | strelka | 2.9.10 |
| TABIX_BGZIPTABIX | tabix | '1.12' |
| TABIX_BGZIPTABIX_INTERVAL_COMBINED | tabix | '1.12' |
| TABIX_BGZIPTABIX_INTERVAL_SPLIT | tabix | '1.12' |
| TABIX_BGZIP_TIDDIT_SV | tabix | '1.12' |
| TABIX_TABIX | tabix | '1.12' |
| TIDDIT_SV | tiddit | 3.6.1 |
| VCFTOOLS_TSTV_COUNT | vcftools | 0.1.16 |
| Workflow | Nextflow | 23.10.1 |
| Workflow | nf-core/sarek | 3.4.0 |
| ExcludeMnps | bcftools | 1.19 |
| splitMultiAllelics | bcftools | 1.19 |
| importGVCF | gatk | 4.5.0.0 |
| genotypeGVCF | gatk | 4.5.0.0 |
| vep | ensembl-vep | 111.0 |
| tabix | htslib | 1.19 |
| variantRecalibrator | gatk | 4.5.0.0 |
| applyVQSRIndel | gatk | 4.5.0.0 |
Updated about 1 year ago
Did this page help you?